The regenerative potential of somatic stem cells declines with age. Our subproject within the FWF-funded SFB F78 (“Neuro Stem Modulation”; Coordinator: Dr. Jürgen Knoblich) aims at deciphering the transcriptional network of neural stem cell (NSC) decline and gain of quiescence to understand regenerative processes in the adult brain. Since experimental access to young vs. old human brain tissue is limited, we employ reprogramming technology as a proxy to analyze physiologic neural regeneration. We use iN- and iNSC-type direct conversion from isogenic sources to gain human NSCs of distinct age. We assess reprogramming trajectories during direct conversion of human fibroblasts into induced NSCs by serial combinatorial cell-indexing. This will enable for parallel capture of clonal history and cell identity to reconstruct multilevel lineage trees as a proxy for regeneration pathways. Moreover, we will perform a CRISPR/Cas-based perturbation screen to study the function of human-specific instructors of neurodevelopment.

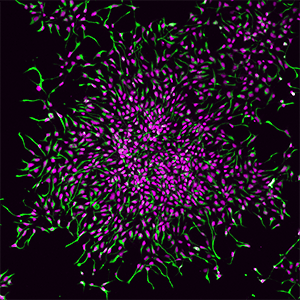
Immunostaining of induced neural stem cells


Precise regulation of intracellular calcium is essential for neuronal activity and network function in the human brain. Voltage-gated calcium channels, such as Cav1.3, tightly control calcium influx and play key roles during both early neural development and in the adult brain. Mutations in Cav1.3 alter channel properties and are associated with a spectrum of neurodevelopmental disorders, including developmental delay, epilepsy, and autism spectrum disorder (ASD). Notably, different pathogenic variants can differentially affect channel function, resulting in highly diverse clinical presentations. A central aim of our research is to understand how variant-specific alterations in Cav1.3 function translate into distinct disease phenotypes. To address this, we use human stem cell–based 2D and 3D neural models to investigate the underlying cellular and molecular mechanisms. Using genome engineering approaches such as prime editing, we generate isogenic induced pluripotent stem cell (iPSC) lines carrying clinically relevant Cav1.3 variants. These cells are differentiated into neural progenitors, neurons, and cortical organoids, enabling the study of human neurodevelopment across multiple stages in vitro and allowing detailed analysis of genotype–phenotype relationships. We combine immunofluorescence, transcriptomic profiling, and calcium imaging to systematically assess the impact of these mutations on cellular function and neural development. Ultimately, our goal is to uncover the mechanisms driving disease phenotypes and to evaluate whether mutation-specific dysfunctions can be mitigated through targeted therapeutic strategies

Structure of a voltage-gated calcium channel.
Batten disease, a form of neuronal ceroid lipofuscinosis (NCL), is a rare pediatric neurodegenerative lysosomal storage disorder. It is characterized by the accumulation of ceroid lipofuscin in the brain, leading to progressive vision loss, seizures, dementia, and a decline in cognitive and motor functions. The disease is caused by mutations in CLN3, a gene encoding a putative lysosomal membrane protein; however, its precise biological function remains poorly understood. With generous support from the NCL Foundation (www.ncl-stiftung.de), we aim to elucidate the pathogenic mechanisms underlying CLN3-associated Batten disease to enable for the development of targeted therapeutic strategies for NCL patients. Our research focuses on early neurodevelopmental defects using human iPSC-derived neurons and cerebral organoids. By employing an allelic series of CLN3 variant iPSCs, we identify early molecular and cellular alterations associated with disease severity. Through comparative analyses of cellular and lysosomal phenotypes across different genotypes and developmental stages, we define early neuronal dysfunction driven by CLN3 mutations
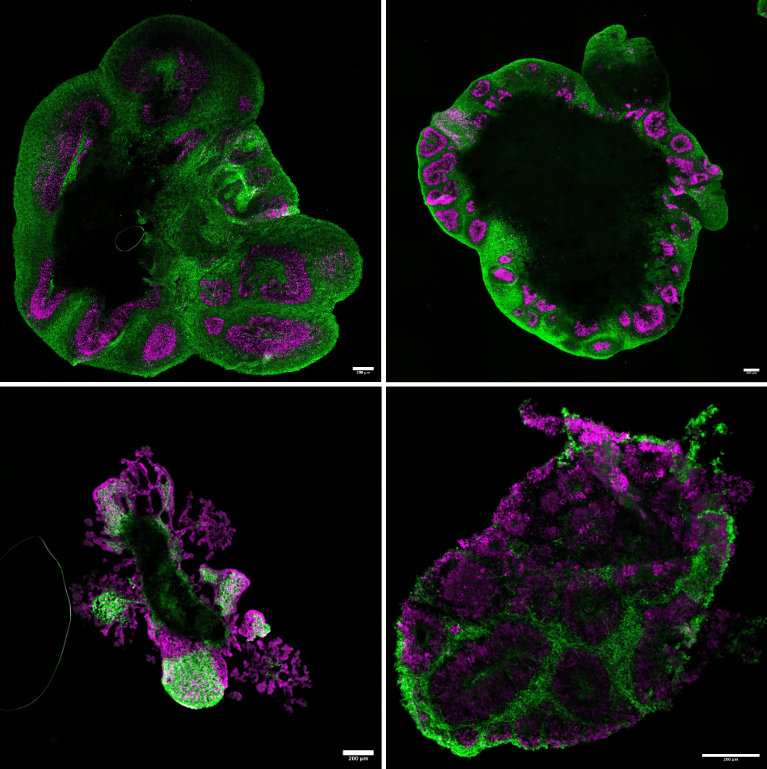
iPSC-derived cortical organoids from CLN3-variants

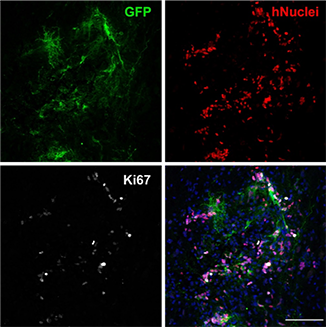
Human NSCs after transplantation into injured rat spinal cords
Induced neural stem cells (iNSCs) carry a strong potential for clinical application. Recently, we demonstrated that iNSCs can ameliorate pathophysiology of multiple sclerosis in a relevant mouse model. Within a cooperative project with the Paracelsus Medical University, Salzburg funded by the wings for life foundation we assess the therapeutic capacity of iNSC to cure traumatic spinal cord injury (SCI). We will apply a combination of anti-inflammatory intervention at the acute phase with transplantation of iNSCs during the chronic phase to induce and maintain a neuroregenerative environment. Stem cells of the nervous system do represent an ideal cellular source for cell replacement; however, the access to appropriate cells from adult patients is limited. By employing patient-specific iNSCs we will make use of a novel, safe and autologous cellular source for transplantation in the chronic phase. Our previous studies have shown that iNSCs can be generated much faster and are safer than iPSC-derived neural cells. However, iNSCs can be cryopreserved, proliferate virtually indefinitely and are able to mature into neuronal and glial cells.
Human NSCs after transplantation into injured rat spinal cords
Establishing an authentic human disease model of the lysosomal storage disease (LSD) Mucopolysaccharidosis IIIB (MPS IIIB), often referred to as Sanfilippo Syndrome, is the goal of this project. Using cellular reprogramming of patient-derived somatic cells, we aim to better understand the cellular dysfunction as well as to develop novel therapeutic approaches for MPS IIIB. In detail, we will investigate the contribution of lysosomal pathway dysfunction to neurodegeneration employing models of the brain and eye using patient-derived reprogrammed cells. By applying specific differentiation protocols, we will generate and analyze patient-specific adherent neuronal networks, retinal cells as well as cortical organoids. By the means of state-of-the-art analytic methods, we will explore to which extent the disease is recapitulated in a petri dish and consequently enhance the understanding of the molecular hallmarks and disease mechanisms of MPS IIIB. The collaboration with Nicole Muschol from UKE, Hamburg, Germany enables us to assess samples from MPS IIIB patients enrolled in a clinical study. Thus, allowing a direct patient-specific analysis and eventually the prediction of therapy efficacy. We anticipate major advances in MPS IIIB disease understanding in particular and rare neurological diseases in general to facilitate clinical translation to novel therapeutic cures.

Disease modelling of Mucopolysaccharidosis type IIIb.


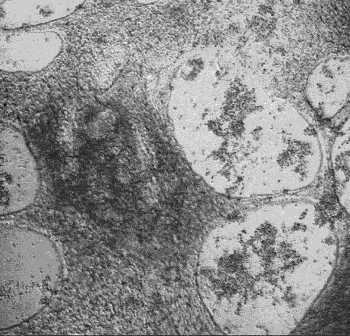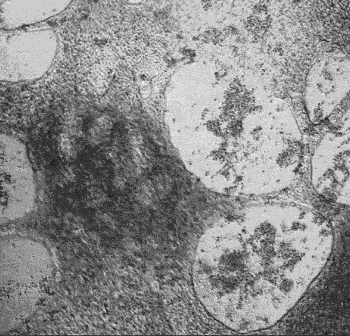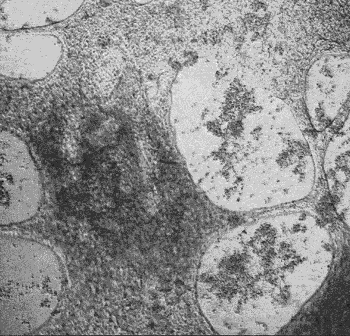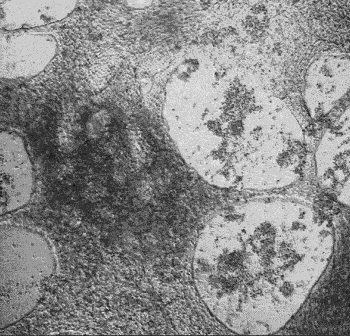
iPSC-derived cardiomyocytes beating in vitro.
Ageing is a major risk factor for cardiovascular disease. At organismic level cardiovascular ageing is characterized by a gradual change of vascular structure and cardiac homeostenosis resulting in vascular and cardiac insufficiency. Within the FFG-funded COMET center VASCage we aim to get a further understanding of cardiovascular ageing at cellular and molecular level. Employing stem cells we will determine cardiovascular ageing profiles with high predictive power and try to identify putative molecular targets for therapeutic intervention. Current research is limited by the poor accessibility to functional human tissues. Human pluripotent stem cells (PSCs) offer a virtually unlimited access to functional human cells to model cardiovascular ageing. We generate hiPSCs from patients suffering from cardiomyopathy and stroke, respectively, employ robust in vitro differentiation into cardiovascular lineages and develop human 3D in vitro models for myocard and vascular structures. We will determine genome-wide ageing profiles by omics technologies defined by both physiological and synthetic, premature ageing.
iPSC-derived cardiomyocytes beating in vitro.
Parkinson’s Disease (PD) is a neurodegenerative disease characterized by a progressive loss of striatal dopaminergic (DA) projecting neurons of the substantia nigra. The pathological hallmarks of PD are intracellular inclusions of proteins called Lewy bodies and Lewy neurites that are predominantly composed of misfolded and aggregated forms of the presynaptic protein alpha-Synuclein (a-Syn), encoded by the SNCA gene. This neuronal degeneration leads to marked decrease of dopamine levels in the striatum, which triggers a range of motor deficits characterising PD as a movement disorder such as tremor at rest, rigidity and bradykinesia. Recent studies indicate important roles of synuclein proteostasis, mitochondrial function, neuroinflammation and ageing in PD pathogenesis. To analyse these processes in relevant human cells we generate novel human iPSC-derived 3D model systems in vitro. Human models will be derived from genetic and idiopathic PD iPSC lines, subjected to premature ageing and a-Syn aggregation and pro-inflammatory cytokine production will be analysed.
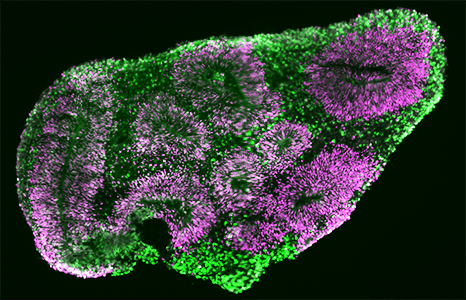
Section of a brain organoid at 4 weeks, showing neural progenitors organized in rosettes.